
Protein Folding
Protein folding is the physical process by which a linear polypeptide folds into its characteristic and functional three-dimensional structure. Folding of a polypeptide chain is strongly influenced by the solubility of the AA R-groups in water. Each protein exists as an unfolded polypeptide or random coil when translated from a sequence of mRNA to a linear chain of amino acids. This polypeptide lacks any stable (long-lasting) three-dimensional structure (the left hand side of the neighboring figure). Amino acids interact with each other to produce a well-defined three-dimensional structure, the folded protein (the right hand side of the figure), known as the native state. All the information for the native fold appears therefore to be contained within the primary structure (Anfinsen received the Nobel Prize for this), and proteins are self-folding (although in vivo, polypeptide folding is often assisted additional molecules known as molecular chaperones).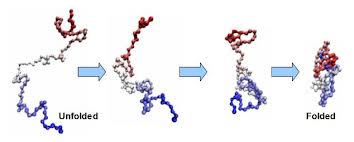
Minimizing the number of hydrophobic side-chains exposed to water (the hydrophobic effect) is an important driving force behind the folding process. Intramolecular hydrogen bonds also contribute to protein stability (think of their importance in secondary structures). Ionic interactions (attraction between unlike electric charges of ionized R-groups) also contribute to the stability of tertiary structures. Disulfide bridges (covalent bonds) between neighboring cysteine residues can also stabilize three-dimensional structures. Note that disulfide bonds are rarely observed in intracellular proteins because of the reducing intracellular environment.
The correct 3D structure of a protein is essential to its function, although some parts of functional proteins may remain unfolded. Failure to fold into native structure generally produces inactive proteins, but in some instances misfolded proteins have modified or toxic functionality (think prions & amyloid fibrils). Consistent with their functional importance, three-dimensional structures of proteins are more conserved during evolution time than are the primary amino-acid sequences.
For those who want to contribute to science while playing games I suggest you check out FoldIt. Recently players of this game were able to correctly predict the structure of a retroviral protease. For those who want their spare CPU cycles to go to a good use, I suggest you check out Folding@home.