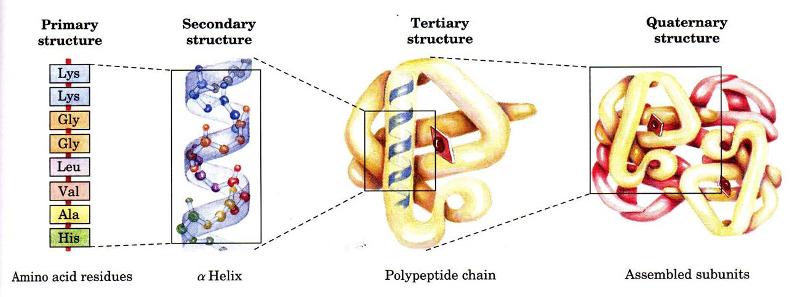
Levels of Protein Organization
A 2014 Foundations of Medicine eLAB
A protein's primary structure is defined as the amino acid sequence of its polypeptide chain; secondary structure is the local spatial arrangement of a polypeptide's backbone (main chain) atoms; tertiary structure refers to the three-dimensional structure of an entire polypeptide chain; and quaternary structure is the three-dimensional arrangement of the subunits in a multisubunit protein. In this series of pages we examine the different levels of protein organization. We also view structures in lots of ways -- Cα backbone, ball-and-stick, CPK, ribbon, spacefilling -- as well color is used to highlight different aspects of the amino acids, structure, etc. As you traverse though this module please note these aspects.
 This module includes links to KiNG (Kinemage, Next Generation), which displays three-dimensional structures in an animated, interactive format. These "kinemages" (kinetic images) can be rotated, moved, and zoomed, and parts can be hidden or shown. Kinemages were originally implemented under the auspices of the Innovative Technology Fund and the Protein Society, and the programming and maintenance are done by David C. Richardson and Jane S. Richardson.
This module includes links to KiNG (Kinemage, Next Generation), which displays three-dimensional structures in an animated, interactive format. These "kinemages" (kinetic images) can be rotated, moved, and zoomed, and parts can be hidden or shown. Kinemages were originally implemented under the auspices of the Innovative Technology Fund and the Protein Society, and the programming and maintenance are done by David C. Richardson and Jane S. Richardson.
Reference: "THE KINEMAGE: A TOOL FOR SCIENTIFIC COMMUNICATION" D.C. Richardson and J.S. Richardson (1992) Protein Science 1: 3-9. Also Trends in Biochem. Sci. (1994) 19: 135-8.
Text adapted from: Demo5_4a.kin
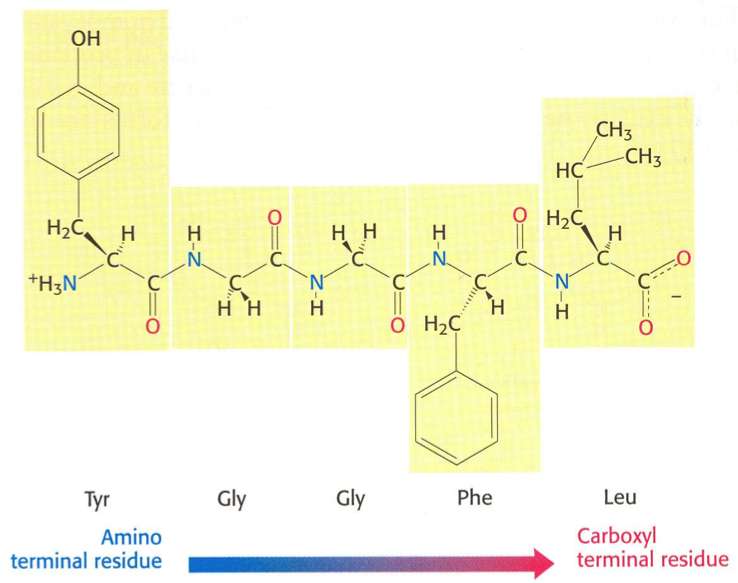
The primary structure of a peptide or protein is the linear sequence of its amino acids (AAs). By convention, the primary structure of a protein is read and written from the amino-terminal (N) to the carboxyl-terminal (C) end. Each amino acid is connected to the next by a peptide bond.
While primary structure describes the sequence of amino acids forming a peptide chain, secondary structure refers to the local arrangement of the chain in space. Several common secondary structures have been identified in proteins. These will be described in the following sections and visualized using the KiNG software mentioned previously.
To load the KiNG Java Applet, just click here. Upon loading this page, the KiNG Java Applet should automatically spawn. If you need information on using King, please hover here.
 An alpha helix is an element of secondary structure in which the amino acid chain is arranged in a spiral. The kinemage linked above shows an individual alpha helix, viewed from the N-terminal end to resemble the "helical wheel" (see figure below). The O and N atoms of the helix main chain are shown as red and blue balls, respectively. The non-integral, 3.6-residue-per-turn repeat of the alpha helix means that the Cα's of successive turns are about halfway offset, giving the main chain a distinctive 7-pointed star appearance in end view. Notice that the Cα-Cβ bonds do not point out radially from the helix axis but "pinwheel" along the line of one of the adjacent peptides, giving the side chains an asymmetrical start.
An alpha helix is an element of secondary structure in which the amino acid chain is arranged in a spiral. The kinemage linked above shows an individual alpha helix, viewed from the N-terminal end to resemble the "helical wheel" (see figure below). The O and N atoms of the helix main chain are shown as red and blue balls, respectively. The non-integral, 3.6-residue-per-turn repeat of the alpha helix means that the Cα's of successive turns are about halfway offset, giving the main chain a distinctive 7-pointed star appearance in end view. Notice that the Cα-Cβ bonds do not point out radially from the helix axis but "pinwheel" along the line of one of the adjacent peptides, giving the side chains an asymmetrical start.
The hydrophobic side chains are shown in seagreen, polar ones in skyblue, and charged ones in red. These can be turned on by clicking on the checkbox labeled "side ch". Now TURN ON and OFF the various display groups and sets, by clicking in the appropriate button box.
When you clicked the different sidechain types on, what did you observe? Did you notice that the helix has one side with mainly polar residues, and the other with mainly hydrophobic residues?. This is a typical globular-protein helix; in its native configuration, the polar residues would face the solvent while the hydrophobic residues would face the protein interior. In the view menu in KiNG, choose View2 or View3 to see more of the structure.
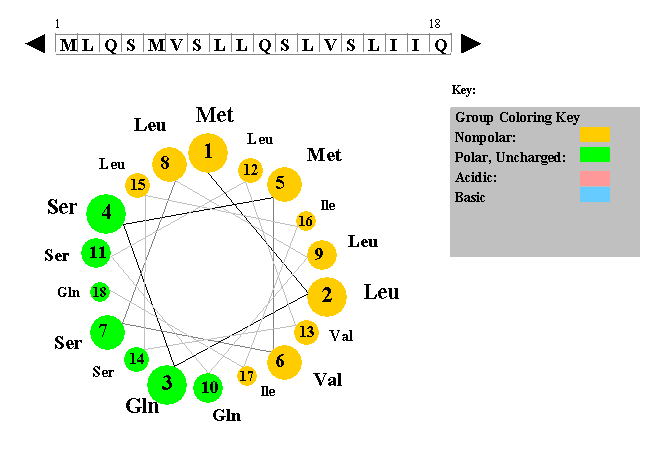 The figure to the left shows a helical wheel representation of an amino acid sequence, as if looking down the axis of an alpha helix that is perpendicular to the page. The amino acid residues are numbered from nearest to most distant and are arranged as an ideal alpha helix with 3.6 residues per complete turn. This figure is a snaphot of a Java Applet written by Edward K. O'Neil and Charles M. Grisham (University of Virginia in Charlottesville, Virginia).
The figure to the left shows a helical wheel representation of an amino acid sequence, as if looking down the axis of an alpha helix that is perpendicular to the page. The amino acid residues are numbered from nearest to most distant and are arranged as an ideal alpha helix with 3.6 residues per complete turn. This figure is a snaphot of a Java Applet written by Edward K. O'Neil and Charles M. Grisham (University of Virginia in Charlottesville, Virginia).
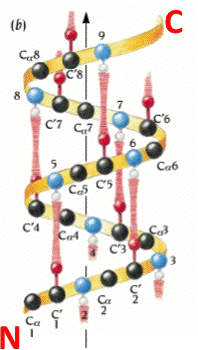
In KiNG, choose View4 for a close-up from the side, with the helical hydrogen bonds (H-bonds) in brown. Turn on "Hbonds" on the button panel, to see the H-bonds in brown. Click on backbone atoms at either end of one of the H-bonds, to verify that the alpha-helical H-bond pattern does indeed go from a donor NH at residue i to an acceptor O at residue i-4 (as shown in the figure to the right). Check to see if this alpha helix has 3.6 residues per turn. If you were to mesure, the rise of a full turn is 5.4 Angstroms (Â).
Alpha helices are nearly all right-handed. To see that this one is righthanded, hold your right hand with the thumb pointing up and the fingers loosely curled; trying to match the spiral of the helix, move slowly along the direction your thumb points and curl along the line of your fingers, as though tightening a screw. When that motion matches the backbone spiral if done with the right hand, then the helix is righthanded.
 To measure phi,psi angles for the KiNG example helix, turn on "Measure angle & dihedral" on the "Tools" pulldown menu. Start by clicking on a carbonyl C atom near the top, then the next N, then the Cα, and then a C again; at that point the information line will show a dihedral angle that is the phi angle of the central N-Cα bond of those 4 atoms. For a righthanded alpha-helix, it should be in the range of -50 to -80 degrees. Click on the next N and you will get the psi angle, which should be between -25 and -60 degrees. Continue down the helix backbone, getting omega (near 180 degrees), phi, psi, etc. These helical phi,psi values are in the well-populated area in the lower left of the Ramachandran plot (shown on the right).
To measure phi,psi angles for the KiNG example helix, turn on "Measure angle & dihedral" on the "Tools" pulldown menu. Start by clicking on a carbonyl C atom near the top, then the next N, then the Cα, and then a C again; at that point the information line will show a dihedral angle that is the phi angle of the central N-Cα bond of those 4 atoms. For a righthanded alpha-helix, it should be in the range of -50 to -80 degrees. Click on the next N and you will get the psi angle, which should be between -25 and -60 degrees. Continue down the helix backbone, getting omega (near 180 degrees), phi, psi, etc. These helical phi,psi values are in the well-populated area in the lower left of the Ramachandran plot (shown on the right).
In summary, the ideal alpha helix has the following properties:
Some general properties of alpha-helices:
Some text adapted from: Kinemage Supplement to Branden & Tooze "Introduction to Protein Structure", Chapter 2 - MOTIFS OF PROTEIN STRUCTURE by Jane S. and David C. Richardson.
A beta strand is an element of secondary structure in which the protein chain is nearly linear. Adjacent beta strands can hydrogen bond to form a beta sheet (also referred to as a beta pleated sheet). The participating beta strands are not continuous in the primary sequence, and do not even have to be close to each other in the sequence, i.e. the strands forming a beta sheet can be separated in primary structure by long sequences of amino acids that are not part of the sheet. Approximately a quarter of all residues in a typical protein are in beta strands, though this varies greatly between proteins
To view a beta sheet in the KiNG Java Applet, click here. Kinemage 1 shows the 6-stranded parallel beta sheet from domain 1 of lactate dehydrogenase (file 1LDM). This doubly-wound parallel beta sheet is the most common folding pattern found in known protein structures. This "fold" is also known as the "nucleotide-binding domain", because most examples bind a mononucleotide (such as FMN) or a dinucleotide (such as NAD) near the middle of one end of the beta sheet. Lactate dehydrogenase is the classic, first-seen example of this type of structure and has the most frequently-observed topology of beta connections.
Notice that the H-bonds in this parallel shet are slanted in alternate directions, rather than perpendicular to the strands as we will see in antiparallel sheets. Drag right or left to better see that the sheet as a whole twists. This twist is usually described by the twist in orientation of the peptide planes (or H-bond plane) as one progresses along the strand; by this definition beta sheet twist is always right-handed, although by varying amounts. Click on atoms along a strand to tell its direction from the residue numbers, and satisfy yourself that all six strands are indeed parallel. The strand labels show strand sequence order. Note that most sequential pairs are next to each other, and that the chain starts in the middle, moves to one edge, skips back to the middle and then moves out to the other edge.There are three possible ways to form a beta sheet from beta strands, discussed below.
1) Parallel beta sheet - All bonded strands have the same N to C direction. As a result they have to be separated by long sequence stretches. The hydrogen bonds are equally distanced.
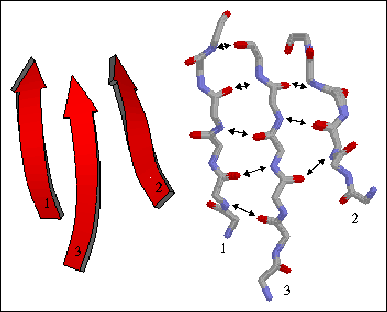
The figure to the left shows a three-stranded parallel beta sheet from the protein thioredoxin. The three parallel strands are shown in both cartoon format (left) and in stick form containing backbone atoms N, CA, C, and O' (right). Hydrogen bonds are identified by arrows connecting the donor nitrogen and acceptor oxygens. Strands are numbered according to their relative position in the polypeptide sequence.
2) Antiparallel beta sheet - The beta strands run in alternating directions and therefore can be quite close on the primary sequence. The distance between successive hydrogen bonds alternates between shorter and longer.
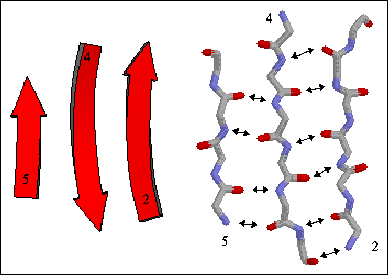
The figure to the right shows a three-stranded antiparallel beta sheet from thioredoxin. The three antiparallel strands are shown in both cartoon format (left) and in stick form containing backbone atoms N, CA, C, and O' (right). Hydrogen bonds are identified by arrows connecting the donor nitrogen and acceptor oxygens. Strands are numbered according to their relative position in the polypeptide sequence.
3) Mixed beta sheet - a mixture of parallel and antiparallel hydrogen bonding. About 20% of all beta sheets are mixed.
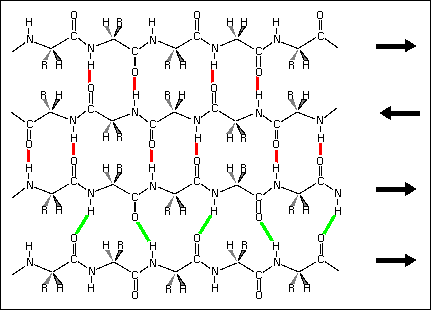
Hydrogen bond patterns in a mixed beta sheet (figure to the left). Here a four-stranded beta sheet containing three antiparallel strands and one parallel strand is drawn schematically. Hydrogen bonds between antiparallel strands are indicated with red lines, those between parallel strands with green lines.
Some of the main features of beta sheets include:
Turn on the side chains in KiNG to examine their arrangment. Along a given strand the sidechains alternate between one side of the sheet (gold) and the other (sea or sky). On adjacent strands the alternation is in register, so that the side chains form rows that are in quite close contact. On parallel beta sheet, the geometry is such that sidechains with branched beta-carbons (Val, Ile, or Thr) make quite favorable contact along a row; since these positions are usually buried and hydrophobic, the result is that Val and Ile are the dominant residues found in these positions. The edge strands, or the very ends of a given strand, can be exposed to solvent and often have significantly more hydrophilic residues (as, for instance, in row 0 here, or the Ser on strand 3).
Some text adapted from: "The Protein Tourist: DOUBLY-WOUND PARALLEL ALPHA/BETA PROTEINS, OR NUCLEOTIDE-BINDING DOMAINS" by J.S. Richardson and D.C. Richardson.
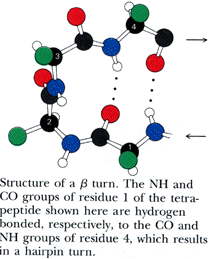 Turns generally occur when the protein chain needs to change direction in order to connect two other elements of secondary structure. The most common is the beta turn, in which the change of direction is executed in the space of four residues. Some commonly observed features of beta turns are a hydrogen bond between the C=O of residue i and the N-H of residue i+3 (i.e, between the first and the fourth residue of the turn) and a strong tendency to involve glycine and/or proline. You will sometimes hear the phrase "beta hairpin" which can be used to describe a beta turn joining two anti-parallel beta strands together. Beta turns are subdivided into numerous types on the basis of the details of their geometry.
Turns generally occur when the protein chain needs to change direction in order to connect two other elements of secondary structure. The most common is the beta turn, in which the change of direction is executed in the space of four residues. Some commonly observed features of beta turns are a hydrogen bond between the C=O of residue i and the N-H of residue i+3 (i.e, between the first and the fourth residue of the turn) and a strong tendency to involve glycine and/or proline. You will sometimes hear the phrase "beta hairpin" which can be used to describe a beta turn joining two anti-parallel beta strands together. Beta turns are subdivided into numerous types on the basis of the details of their geometry.
Gamma turns are three-residue turns which often incorporate a hydrogen bond between the C=O of residue i and the N-H of residue i+2.
Some regions of the protein chain do not form regular secondary structure and are not characterized by any regular hydrogen bonding pattern. These regions are known as random coils and are found in two locations in proteins:
Random coils can be 4 to 20 residues long, although most loops are not longer than 12 residues. Most loops are exposed to the solvent and are have polar or charged side-chains. In some cases loops have a functional role, but in many cases they do not. As a result, loop regions are often poorly conserved (i.e. more prone to change) during evolution.
Some text adapted from: "EXERCISE 3. PROTEIN SECONDARY STRUCTURES" by Kim M. Gernert and Kim M. Kitzler.
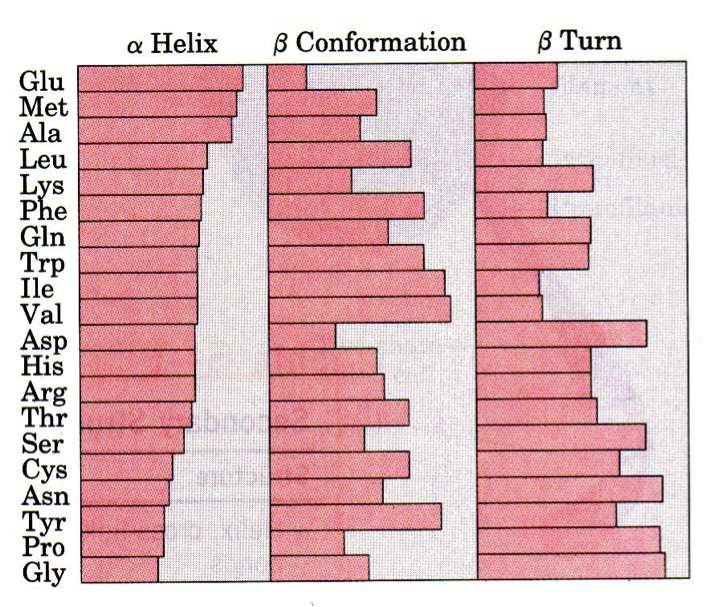
As we have learned, the order of the AAs is the primary structure and all residues in a polypeptide chain have the same main-chain atoms. What vary are the side chains (R groups). Do the specific AAs present dictate the secondary structure? As shown in the figure, all amino acids can be found in all secondary structure elements, but some are more or less common in certain elements. Pro and Gly, for isntance, aren't good in helices but are favored in beta-turns. If we take this a step further and ask whether 2, 3, or 4 amino acids combinations dictate secondary structure we find a stronger correlation, but still not strong enough to reliably predict tertiary structure.
Proteins are abundant in all organisms and are fundamental to life. The diversity of protein structure underlies the very large range of their functions: enzymes (biological catalysts), storage, transport, messengers, antibodies, regulation, and structural proteins.
Proteins are linear heteropolymers of fixed length; i.e. a single type of protein always has the same number and composition of AAs, but different proteins may have 100 to more than 1000 AAs. There is therefore a great diversity of possible protein sequences. The linear chains fold into specific three-dimensional conformations, which are determined by the sequence of amino acids and therefore are also extremely diverse, ranging from completely fibrous to globular. Covalent disulfide bonds can be introduced between cysteine residues placed in close proximity in 3D space -- this provides rigidity for the resulting 3D structure. Ribbon diagrams like the one shown here are a common way to visualize proteins.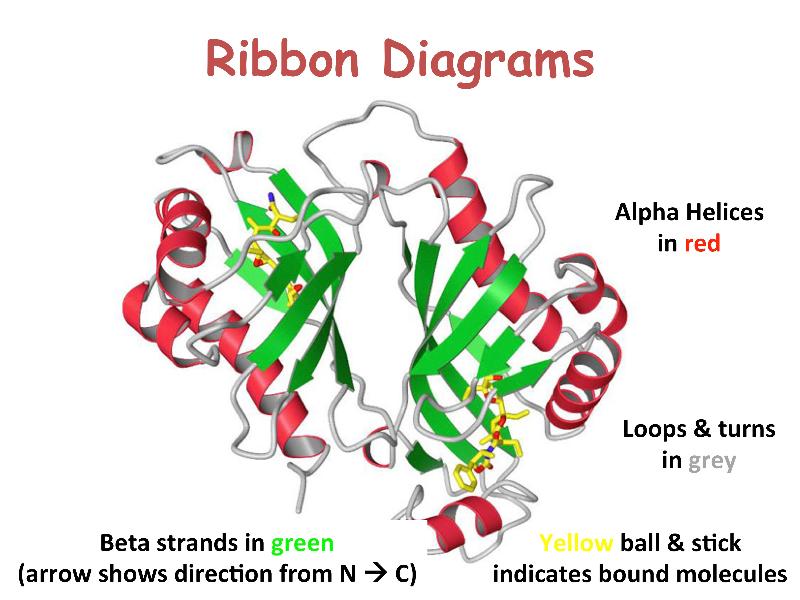
Protein structures can be determined to an atomic level by X-ray diffraction and neutron-diffraction studies of crystallized proteins, and more recently by nuclear magnetic resonance (NMR) spectroscopy of proteins in solution. The structures of many proteins, however, remain undetermined.
To view an example of tertiary structure in KiNG, click here. This is ribonuclease A, an enzyme responsible for the degradation of RNA. The image depicts all atoms of one half of the molecule (cyan for side chains, brown for hydrogen atoms) and just main chain and side chains for the other half. The alternate view shows main-chain atoms and H-bonds (purple). Click "Animate" to cycle between the views.
Although hydrogens constitute about half the atoms in a protein, they are seldom shown explicitly because they are hard to detect with x-ray crystallography (due to low electron density) and they very much complicate the picture. This ribonuclease image is a joint x-ray/neutron diffraction structure, for which hydrogens are always included. Even without H atoms, an all-atom view is too crowded to be very useful but is a good way to appreciate where simplified versions start from.
Some text adapted from: Kinemage Supplement to Branden & Tooze "Introduction to Protein Structure", Chapter 2 - MOTIFS OF PROTEIN STRUCTURE, Jane S. and David C. Richardson.
Protein folding is the physical process by which a linear polypeptide folds into its characteristic and functional three-dimensional structure. Folding of a polypeptide chain is strongly influenced by the solubility of the AA R-groups in water. Each protein exists as an unfolded polypeptide or random coil when translated from a sequence of mRNA to a linear chain of amino acids. This polypeptide lacks any stable (long-lasting) three-dimensional structure (the left hand side of the neighboring figure). Amino acids interact with each other to produce a well-defined three-dimensional structure, the folded protein (the right hand side of the figure), known as the native state. All the information for the native fold appears therefore to be contained within the primary structure (Anfinsen received the Nobel Prize for this), and proteins are self-folding (although in vivo, polypeptide folding is often assisted additional molecules known as molecular chaperones).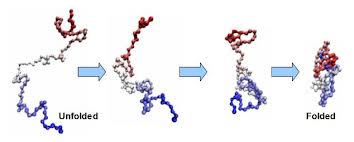
Minimizing the number of hydrophobic side-chains exposed to water (the hydrophobic effect) is an important driving force behind the folding process. Intramolecular hydrogen bonds also contribute to protein stability (think of their importance in secondary structures). Ionic interactions (attraction between unlike electric charges of ionized R-groups) also contribute to the stability of tertiary structures. Disulfide bridges (covalent bonds) between neighboring cysteine residues can also stabilize three-dimensional structures. Note that disulfide bonds are rarely observed in intracellular proteins because of the reducing intracellular environment.
The correct 3D structure of a protein is essential to its function, although some parts of functional proteins may remain unfolded. Failure to fold into native structure generally produces inactive proteins, but in some instances misfolded proteins have modified or toxic functionality (think prions & amyloid fibrils). Consistent with their functional importance, three-dimensional structures of proteins are more conserved during evolution time than are the primary amino-acid sequences.
For those who want to contribute to science while playing games I suggest you check out FoldIt. Recently players of this game were able to correctly predict the structure of a retroviral protease. For those who want their spare CPU cycles to go to a good use, I suggest you check out Folding@home.
Quaternary structure in proteins is the most intricate degree of organization still considered a single molecule. To be considered to have quaternary structure, a protein must have two or more peptide chains forming subunits. The subunits can be different or identical, and in most cases they are arranged symmetrically. In general, a protein with two subunits is called a dimer; one with three subunits a trimer; and one with four subunits a tetramer.
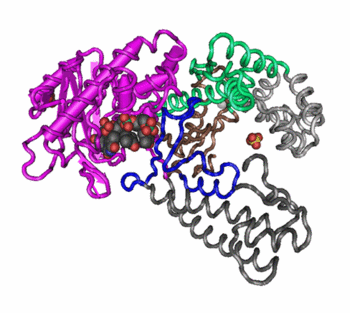 Changes in quaternary structure can occur through conformational changes within individual subunits or through reorientation of the subunits relative to each other. It is through such changes, which underlie cooperativity and allostery in "multimeric" enzymes, that many proteins undergo regulation and perform their physiological function. A good example would be a DNA polymerase (see image) and ion channels. Subunits are held together by the same types of interactions that stabilize the tertiary structure of proteins.
Changes in quaternary structure can occur through conformational changes within individual subunits or through reorientation of the subunits relative to each other. It is through such changes, which underlie cooperativity and allostery in "multimeric" enzymes, that many proteins undergo regulation and perform their physiological function. A good example would be a DNA polymerase (see image) and ion channels. Subunits are held together by the same types of interactions that stabilize the tertiary structure of proteins.
There is debate as to whether quaternary structure should be defined to include peptides linked by covalent (disulfide) bonds. In CMB, we will us quaternary structure to refer only to arrangement of subunits that are not covalently linked, although covalent disulfide bonds may occur within the individual subunits.