Protein Purification Methods
Foundations of Clinical Sciences
It is through protein purification methods that we have been able to study and understand proteins in detail. These methods, or derivatives of the methods, are used in the clinical labs to identify abnormal samples. Protein purification methods use fraction techniques which are in a large part based on:
solubility;
size;
charge;
binding specificity.
These properties of a protein are derived from the AA properties composing the protein. For example the molecular weight (MW) of a protein is just the summation of the masses of the individual AAs composing the protein. MW is usually expressed in daltons (Da) or kilodaltons (kDa). A Da is the same as an atomic mass unit which is approximately the mass of a nucleon and is equivalent to 1 g/mol.
To begin any sort of purification it is important that an assay be available to identify where the protein of interest is after the fractionation. Assays come in many different forms and depends in a large part on the type of protein you are trying to purify (i.e. is it an enzyme?). Commonly used assay technologies are:
spectroscopic (using Bradford reagent or a chromagenic substrate)
immunological (using a antibody that can recognize the protein of interest)
These will be discussed in more detail later.
To being any sort of purification procedure you need to obtain the material from which you plan the isolate the material. Historically the abundance and ease of isolation dictated which proteins were first studied (e.g. hemoglobin). Also many proteins are common to a large number of species (e.g. metabolic enzymes) so they could be isolated in large abundance from other sources, such as yeast or bovine.
Once you have gathered the material containing the protein you want to study it is necessary to generate a crude extract -- for proteins from muscle that would mean grinding it up, for an intercellular protein that would mean breaking the cells open, etc. This is always done in the presence of a buffer and inhibitors. Why? As a scientist you want to control the environment -- keeping the protein you are interested in at a non-denaturing pH, you want to keep it from being cleaved by enzymes that will be released in this process so general inhibitors will be important, etc.
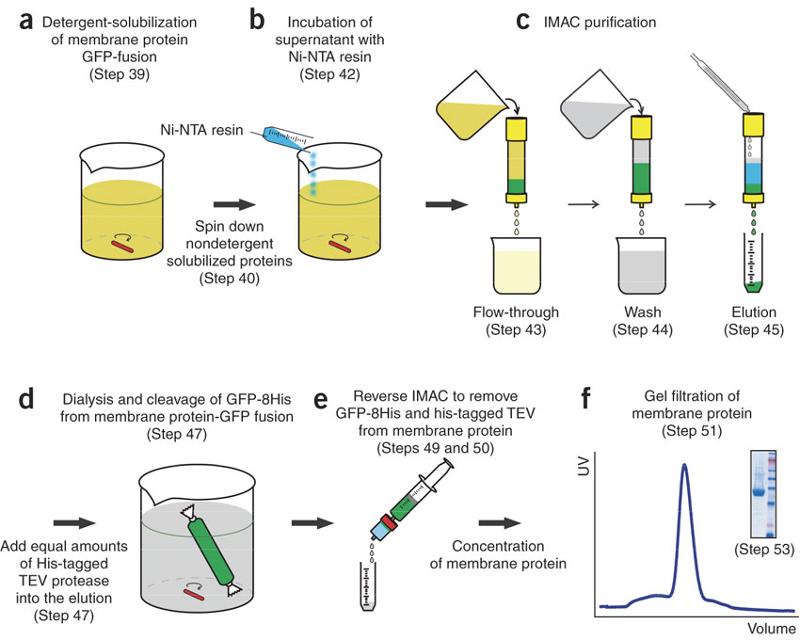
Generally the first step after forming a crude extract is a simple filtration or centrifugation to remove the large material. Centrifugation is a process that involves the use of the centrifugal force for the sedimentation of mixtures with a centrifuge. This process is used to separate two immiscible liquids with more-dense components of the mixture migrate away from the axis of the centrifuge, while less-dense components of the mixture migrate towards the axis. Centrifugation alters the effective gravitational force on to tube/bottle so as to more rapidly and completely cause the precipitate ("pellet") to gather on the bottom of the tube. The remaining solution is properly called the "supernatant". The supernatant liquid is quickly decanted from the tube/bottle without disturbing the precipitate.
Differential centrifugation, as shown in the figure, is multiple rounds of centrifugation at increased speeds and time allows for different cellular fractions to be separated.
At this point, however, an assay is key because is your protein of interest?!
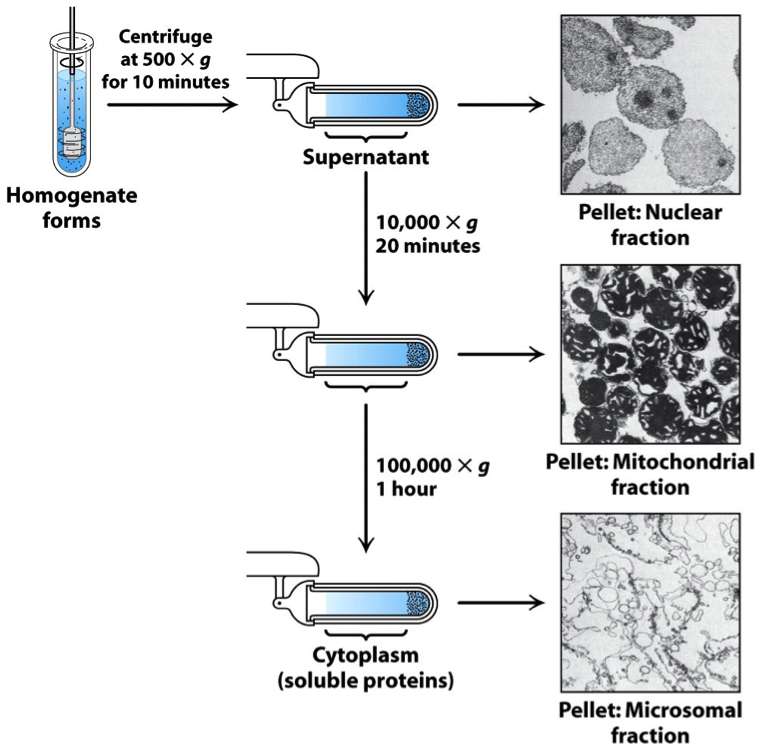
 Dialysis is a procedure for exchanging the solvent around a protein. In general the protein solution is placed inside a semi-permeable membrane (dialysis bag) which is suspended in a larger volume of buffered solution (see image to the right). The key to this procedure working is that the membrane has to be permeable to water and ions, but not to your protein of interest. Thus buffers & salts exchange until an equilibrium is established between the inside & outside of the membrane.
Dialysis is a procedure for exchanging the solvent around a protein. In general the protein solution is placed inside a semi-permeable membrane (dialysis bag) which is suspended in a larger volume of buffered solution (see image to the right). The key to this procedure working is that the membrane has to be permeable to water and ions, but not to your protein of interest. Thus buffers & salts exchange until an equilibrium is established between the inside & outside of the membrane.
Naturally in medicine the types of dialysis you are likely to see are hemodialysis and peritoneal dialysis which remove wastes and excess water from the blood in different ways. Hemodialysis removes wastes and water by circulating blood outside the body through an external filter containing a semipermeable membrane. The blood flows in one direction and the dialysate flows in the opposite. The counter-current flow of the blood and dialysate maximizes the concentration gradient of solutes between the blood and dialysate, which helps to remove more urea and creatinine from the blood. The concentrations of solutes (for example potassium, phosphorus, and urea) are undesirably high in the blood, but low or absent in the dialysis solution, and constant replacement of the dialysate ensures that the concentration of undesired solutes is kept low on this side of the membrane. The dialysis solution has levels of minerals like potassium and calcium that are similar to their natural concentration in healthy blood.
In peritoneal dialysis, wastes and water are removed from the blood inside the body using the peritoneal membrane of the peritoneum as a natural semipermeable membrane. Wastes and excess water move from the blood, across the peritoneal membrane, and into a special dialysis solution, called dialysate, placed in the abdominal cavity. Diffusion and osmosis drive waste products and excess fluid through the peritoneum into the dialysate until the dialysate approaches equilibrium with the body's fluids. Then the dialysate is drained, discarded, and replaced with fresh dialysate often 4-5 times pr day.
Column chromatography is one of the most powerful fractionation methods. It can separate components of mixtures based upon:
size (gel filtration/size exclusion)
charge (ion exchange)
affinity (a specific binding affinity)
Commonalities between all three types of chromatography methods is that they all use a resin (solid phase) with special chemical properties held in a glass cylinder (called a "column"). A buffered solution (mobile phase) percolates through the column and is collected in tubes ("fractions") upon exiting the column. A protein mixture is applied in the mobile phase & percolates through through the column as an expanding band. Different proteins migrate differently depending on their properties and those of the resin.
Below we will examine the different forms and examine their particular properties.
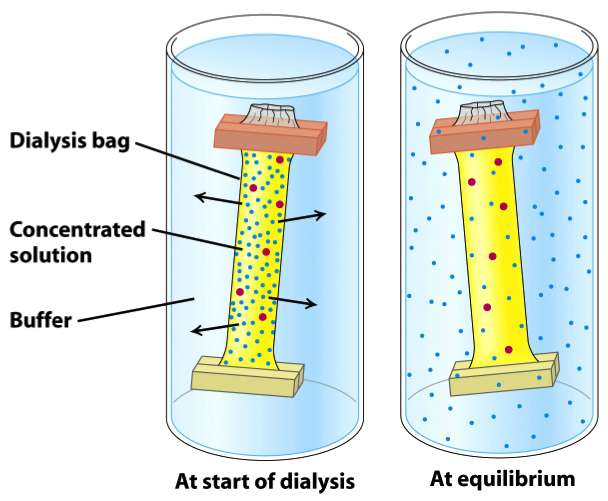
 In gel filtration, or as it is sometimes referred to as size exclusion, chromatography the resin are porous (see figure to the left). Some molecules (blue here) can enter the resin and as the lines try to indicate it is not a straight path through; thus it takes longer for small molecules to traverse the column than large molecules which travel around the outside of the resin. This is highlighted in the figure to the right where big molecules (blue) come off first and smaller molecules (red) later.
In gel filtration, or as it is sometimes referred to as size exclusion, chromatography the resin are porous (see figure to the left). Some molecules (blue here) can enter the resin and as the lines try to indicate it is not a straight path through; thus it takes longer for small molecules to traverse the column than large molecules which travel around the outside of the resin. This is highlighted in the figure to the right where big molecules (blue) come off first and smaller molecules (red) later.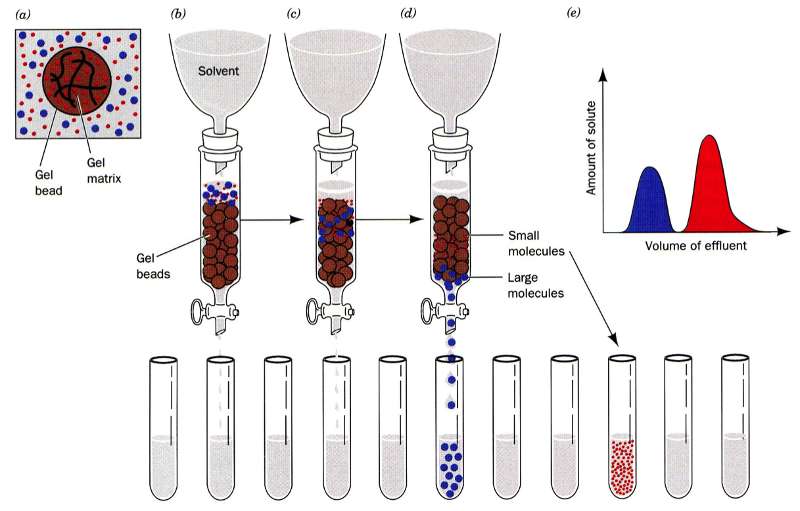
Ion exchange chromatography is broken in to two types - anion & cation exchangers. There are many different types of moieties that are used from weakly to very strongly charged thus allowing a huge range of molecules the ability to interact.
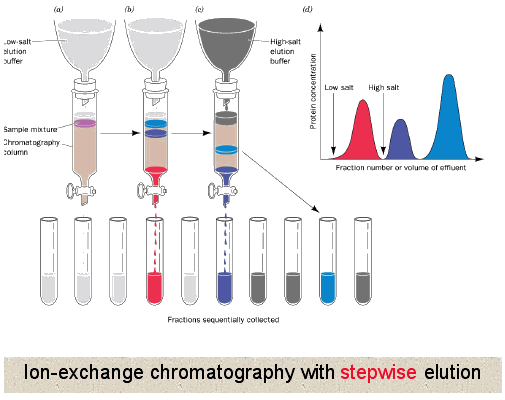
Unlike gel filtration chromatography, here proteins directly interact with the resin. So generally the column is equilibrated in a buffer solution to establish a constant pH in the column, then the protein mixture is loaded where all or some of the proteins interact with the resin depending upon their own charge. Buffer is continued to be applied until all proteins not interacting with the resin have been washed off. At that point usually a gradient of increasing salt concentration (disrupts ionic and hydrogen binding) in the buffer is applied to column allowing the most weakly interacting proteins to release first followed by the more strongly and finally the most strongly interacting. This can also be accomplished by changing the pH of the buffer being applied to the column.
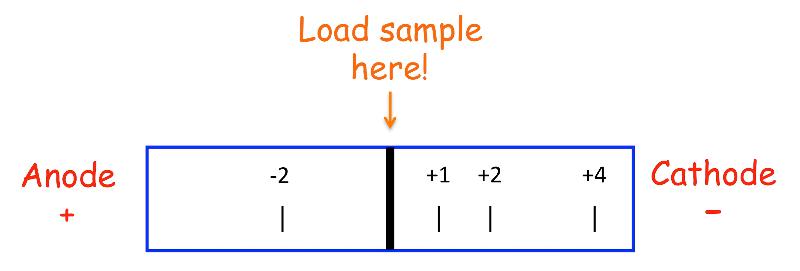
Anion exchanger means that it removes anions from protein mixture so that means the resin must be decorated with positively charged moieties. Before elution begins all positively and uncharged proteins will fall through the column. When you start eluting, first you will knock off the weakly negative proteins (e.g. -1 charge), followed by those with a stronger negative charge (-2), and finally the most negatively charged proteins (-3).
It is exactly oppose with a cation exchanger -- here cations are removed from the protein solution so the resin must be negatively charged. Again before elution begins all negatively and uncharged proteins will fall through the column. When you start eluting, first you will knock off the weakly positive proteins (e.g. +1 charge), followed by those with a stronger positive charge (+2), and finally the most positively charged proteins (+3).
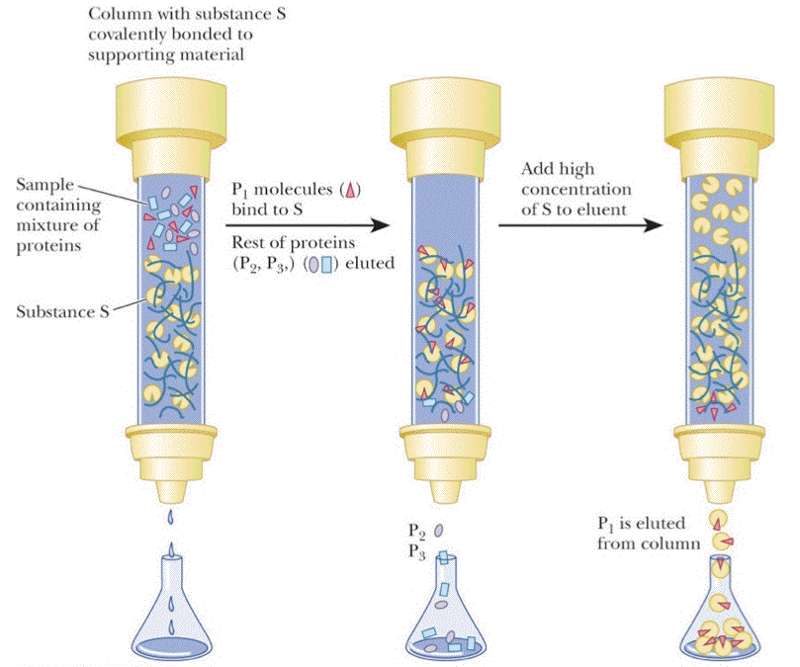
Affinity chromatography requires that you know something specific about your protein -- that it has a specific tag engineered into the sequence, that it binds NAD+, you know the ligand it binds or that you have a specific monoclonal antibody that interacts with your protein. For this type of chromatography your resin is decorated with the antibody, NAD+ or a divalent metal (most popular engineered tag is the 6xHis tag -- 6 His residues at either the N- or C-termini of the protein). Again the column resin is pre-equilibrated in the the appropriate buffer before the protein sample is loaded. It is expected that only the protein of interest will interact. Once all non-interacting proteins have been removed from the column then your protein can be eluted by changing the pH, adding salt, adding a metal chelator, or a high concentration of the ligand.
Electrophoresis is the motion of dispersed particles relative to a fluid under the influence of a uniform electric field. Thus it separates components of a mixture based on their size amd/or charge.
How do you remember which electrode is the cathode and which is the anode?
Simple ... anions travel to the anode and cations travel to the cathode.
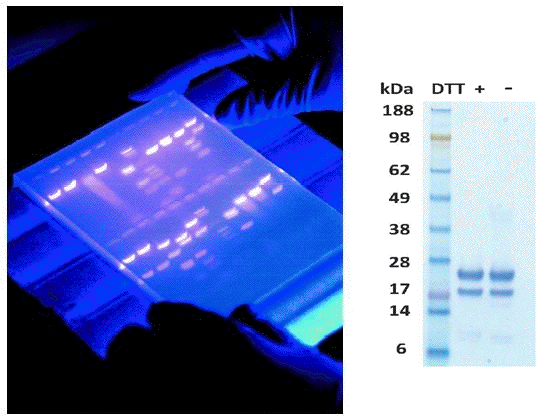
Visualization of proteins on paper or in a gel is an important step in any electrophoresis. Often with DNA the gels are soaked in ethidium bromide which intercalates into DNA and fluoresces under UV light (left image). Proteins may be visualized using silver stain or Coomassie Brilliant Blue dye (right image). In some cases the gels are transferred to a solid support (nitrocellulose) and then probed with specific antibodies (Western Blot).
Generally used to separate AAs or peptides of differing charge. As shown in the figure, AAs and peptides will separate based on their charge with the most highly charged species moving the furthest.
 It is used in clinical chemistry to separate proteins by charge and/or size (IEF agarose, essentially size independent) and in biochemistry and molecular biology to separate a mixed population of DNA and RNA fragments by length, to estimate the size of DNA and RNA fragments or to separate proteins by charge. It is a process which enables the sorting of molecules. Using an electric field, molecules (such as DNA) can be made to move through a gel made of agar or polyacrylamide. The electric field consists of a negative charge at one end which pushes the molecules through the gel, and a positive charge at the other end that pulls the molecules through the gel. The molecules being sorted are dispensed into a well in the gel material. The gel is placed in an electrophoresis chamber, which is then connected to a power source (see figure to the left). When the electric current is applied, the larger molecules move more slowly through the gel while the smaller molecules move faster. The different sized molecules form distinct bands on the gel.
It is used in clinical chemistry to separate proteins by charge and/or size (IEF agarose, essentially size independent) and in biochemistry and molecular biology to separate a mixed population of DNA and RNA fragments by length, to estimate the size of DNA and RNA fragments or to separate proteins by charge. It is a process which enables the sorting of molecules. Using an electric field, molecules (such as DNA) can be made to move through a gel made of agar or polyacrylamide. The electric field consists of a negative charge at one end which pushes the molecules through the gel, and a positive charge at the other end that pulls the molecules through the gel. The molecules being sorted are dispensed into a well in the gel material. The gel is placed in an electrophoresis chamber, which is then connected to a power source (see figure to the left). When the electric current is applied, the larger molecules move more slowly through the gel while the smaller molecules move faster. The different sized molecules form distinct bands on the gel.
The term "gel" in this instance refers to the matrix used to contain, then separate the target molecules. In most cases, the gel is a crosslinked polymer whose composition and porosity is chosen based on the specific weight and composition of the target to be analyzed. When separating proteins or small nucleic acids (DNA, RNA, or oligonucleotides) the gel is usually composed of different concentrations of acrylamide and a cross-linker, producing different sized mesh networks of polyacrylamide. When separating larger nucleic acids (greater than a few hundred bases), the preferred matrix is purified agarose. In both cases, the gel forms a solid, yet porous matrix. Agarose is composed of long unbranched chains of uncharged carbohydrate without cross links resulting in a gel with large pores allowing for the separation of macromolecules and macromolecular complexes.
"Electrophoresis" refers to the electromotive force (EMF) that is used to move the molecules through the gel matrix. By placing the molecules in wells in the gel and applying an electric field, the molecules will move through the matrix at different rates, determined largely by their mass but also their charge and shape which varies widely for proteins. Electrophoretic mobility of small molecules is greater than the mobility of large molecules with the same charge density thus allowing separation. To separate proteins or DNA generally the pH of the buffer and protein mixture is high (~9) so that the proteins carry a net-negative charge. However, because size, charge and shape all play a role in how a molecule will behave in a native gel most scientists use a SDS-PAGE gel which is predictable.
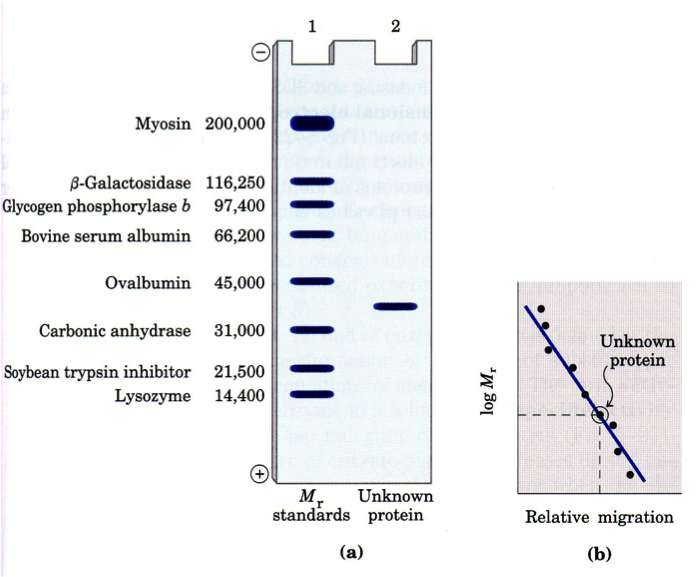 SDS PAGE separate molecules by size because the presence of SDS (sodium dodecyl sulfate) denatures the protein removing 2˚, 3˚ and 4˚ structures (they assume a linear chain) and the SDS coats the molecules giving them a uniform charge/mass ratio. Most often protein samples are also treated with ß-mercaptoethanol (ß-ME) to break any existing disulfide bonds and to give them a linear chain (1˚ structure).
SDS PAGE separate molecules by size because the presence of SDS (sodium dodecyl sulfate) denatures the protein removing 2˚, 3˚ and 4˚ structures (they assume a linear chain) and the SDS coats the molecules giving them a uniform charge/mass ratio. Most often protein samples are also treated with ß-mercaptoethanol (ß-ME) to break any existing disulfide bonds and to give them a linear chain (1˚ structure).
The presence of standards of known size always a calibration curve to be created that can be used to identify the approximate MW of an unknown protein (band).
Isoelectric focusing (IEF) is a technique for separating different molecules by differences in their isoelectric point (pI). It is a type of electrophoresis, usually performed on proteins in a gel, that takes advantage of the fact that overall charge on the molecule of interest is a function of the pH of its surroundings. When an IEF gel is poured a pH gradient is established
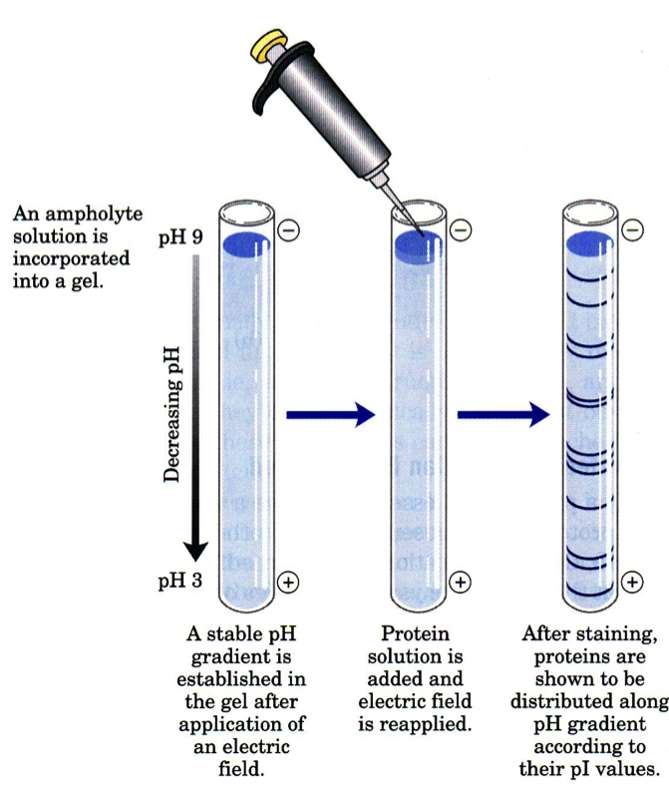
A protein that is in a pH region above its isoelectric point (pI) will be negatively charged and will migrate towards the anode (positive). As it migrates through a gradient of decreasing pH, however, the protein's overall charge will increase until the protein reaches the pH region that corresponds to its pI. At this point it has no net charge and so migration ceases (as there is no electrical attraction towards either electrode). As a result, the proteins become focused into sharp stationary bands with each protein positioned at a point in the pH gradient corresponding to its pI. The technique is capable of extremely high resolution with proteins differing by a single charge being fractionated into separate bands.
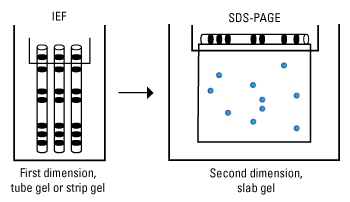
Two-dimensional gel electrophoresis (2D electrophoresis) is a form of gel electrophoresis commonly used to analyze proteins in which mixtures of proteins are separated by two properties in two dimensions on gels. As shown in the figure, 2D electrophoresis begins with an IEF gel (in a tube) which separates proteins based on their pI. This is then laid on top of a SDS-PAGE gel (90 degrees from the first separation). Because it is unlikely that two molecules will be similar in two distinct properties, molecules are more effectively separated in 2D electrophoresis than in 1D electrophoresis.
 Antibodies are proteins made by B cells (part of the body's immune system) with each B cell producing unique antibodies that recognize a specific epitope on the antigen. An antigen is any substance that provokes an immune response – something foreign or toxic to the body. An epitope is a distinct molecular surface of an antigen capable of being bound by an antibody; for proteins these are divided into two categories, conformational epitopes and linear or sequential epitopes, based on their interaction with the antigen. The normal function of an antibody is to bind foreign substances (antigens) and flag them for destruction. This ability of antibodies to recognize and bind an epitope on an antigen makes them an important tool in research and the clinical laboratory. In fact, more than 30 antibodies are currently used therapeutically.
Antibodies are proteins made by B cells (part of the body's immune system) with each B cell producing unique antibodies that recognize a specific epitope on the antigen. An antigen is any substance that provokes an immune response – something foreign or toxic to the body. An epitope is a distinct molecular surface of an antigen capable of being bound by an antibody; for proteins these are divided into two categories, conformational epitopes and linear or sequential epitopes, based on their interaction with the antigen. The normal function of an antibody is to bind foreign substances (antigens) and flag them for destruction. This ability of antibodies to recognize and bind an epitope on an antigen makes them an important tool in research and the clinical laboratory. In fact, more than 30 antibodies are currently used therapeutically.
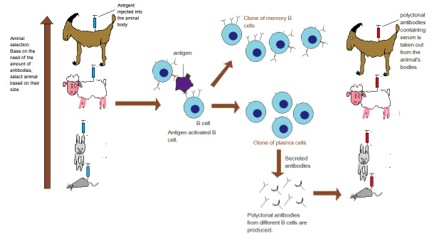
To generate an antibody the antigen (can be a whole protein or fragments of) are injected into an animal (often a mouse, rabbit, goat or donkey) several times over the course of several months. The key is the antigen must be different enough from the animal's own proteins to allow an immune response to be generated.
A polyclonal antibody actually refers to all the antibodies (IgG) that were in the serum of the host at the time the blood was collected. Since we have stimulated the immune system to produce antibodies, lots of B cells will be producing antibodies to many different epitopes on the antigen. Hence why it is called polyclonal.
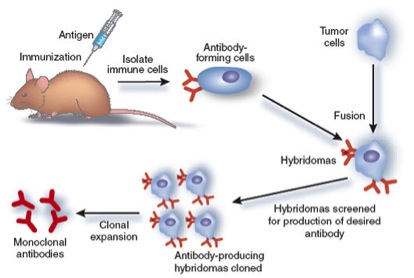
A monoclonal antibody is produced by one cell thus they all recognize the same epitope on the antigen. To produce a monoclonal antibody, tumor cells that can replicate endlessly are fused with B cells from an animal which has been stimulated with an antigen. The result of this cell fusion is a "hybridoma," which will continually produce antibodies.
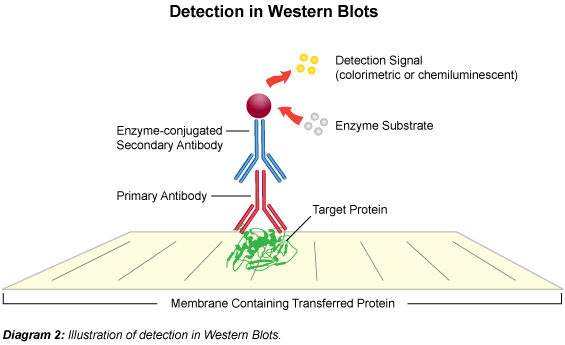 The term "blotting" refers to the transfer of biological samples from a gel to a membrane and their subsequent detection on the surface of the membrane. Western blotting (also called immunoblotting because an antibody is used to specifically detect its antigen) is a routine technique for protein analysis. The specificity of the antibody-antigen interaction enables a target protein to be identified in the midst of a complex protein mixture in a semi-quantitative manner.
The term "blotting" refers to the transfer of biological samples from a gel to a membrane and their subsequent detection on the surface of the membrane. Western blotting (also called immunoblotting because an antibody is used to specifically detect its antigen) is a routine technique for protein analysis. The specificity of the antibody-antigen interaction enables a target protein to be identified in the midst of a complex protein mixture in a semi-quantitative manner.
The first step is to separate the macromolecules using gel electrophoresis (native or SDS-PAGE). After electrophoresis, the separated molecules are transferred to a membrane (usually nitrocellulose). As the membrane will bind any protein (including the antibody you will use to detect your protein of interest), after transferring the sample the membrane must be blocked with a common (cheap!) protein to prevent any nonspecific binding of antibodies to the membrane. Detailed procedures for detection of a protein on a Western blot vary widely. Most laboratories use a indirect detection method, in which a primary antibody is added first to bind to the antigen. This is followed by a labeled secondary antibody which recognizes the primary antibody. Labels include biotin, fluorescent probes, and enzyme conjugates that convert a substrate to a colored product thus staining the membrane.
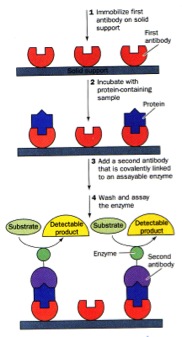 There are MANY forms of ELISAs! Most frequently used is the "sandwich" form in which an antibody is bound to a well in a microtiter dish. The sample is added, incubated, and then protein which were not captured by the antibody washed away. A labeled secondary antibody which recognizes a different part of the bound antigen can be used to quantify the amount of antigen in the sample.
There are MANY forms of ELISAs! Most frequently used is the "sandwich" form in which an antibody is bound to a well in a microtiter dish. The sample is added, incubated, and then protein which were not captured by the antibody washed away. A labeled secondary antibody which recognizes a different part of the bound antigen can be used to quantify the amount of antigen in the sample.
Mass Spectrometry is one of the most important analytical techniques available, and part of the power of the technique lies in its ability to detect minute quantities of material - 100 picograms or less. A mass spectrometer determines the mass of a molecule by measuring the mass-to-charge ratio (m/z) of its ion. Ions are generated by inducing either the loss or gain of a charge from a neutral species. Once formed, ions are electrostatically directed into a mass analyzer, where they are separated according to m/z and finally detected. In general the results of a MS or MS/MS experiment are compared to standards in a database to determine the components of the original sample.